Ecosystem Innovation: IonQ’s Life Sciences Application Workflow Accelerates the Drug Development Process

Innovative Ecosystem Collaboration on a Commercially Relevant Use Case
Quantum computing is moving rapidly from promise to practical impact, and our recent life sciences demonstration is a powerful example of IonQ’s dedication to bringing quantum to the real world. In collaboration with AstraZeneca, AWS, and NVIDIA, we are now ready to share exciting results of the largest quantum-accelerated electronic structure simulation performed to date. This groundbreaking demonstration incorporates state-of-the-art quantum chemistry techniques in an end-to-end, hybrid quantum- and GPU-accelerated workflow to model a critical step in a nickel-catalyzed synthetic chemical reaction, a key process relevant to drug development workflows used at AstraZeneca and across the pharmaceutical industry.
The collaboration integrated IonQ Forte on Amazon Braket, the NVIDIA CUDA-Q framework, and HPC-scale, GPU-accelerated classical postprocessing via AWS ParallelCluster to execute the chemistry simulation at least 20 times faster than our collaborators’ state-of-the-art estimates. This innovative effort demonstrated how tightly-integrated hybrid quantum-classical workflows can potentially enhance accuracy and efficiency in computational chemistry while also improving the time-to-solution and cost-effectiveness of the workflow. With continued advancements and a direct path to scale to hundreds of high-quality, highly connected trapped-ion qubits, this work paves the way for a future where quantum acceleration facilitates faster, more efficient, and more sustainable approaches to drug synthesis and materials science.
Industry Engagement: Bridging Research & Enterprise
At IonQ, working directly with industry customers and partners is core to our business and technical strategy. From the outset, we have believed that there is immense value for both quantum computing providers and enterprise customers in pursuing quantum-enabled innovation together – from discovery to use case identification to application development and beyond, the best results consistently come from relevant quantum and industry experts working side-by-side.
The IonQ applications team, in collaboration with AstraZeneca computational chemists, dedicated extensive time to analyzing computational challenges within the drug discovery and development process, identifying workflows and sub-routines that could be quantum accelerated, and where quantum acceleration would provide meaningful benefit over the classical state of the art.
One well-known challenge in the space is the physical modeling of molecular interactions at increased accuracy, but while molecular simulation is widely recognized as a promising application for quantum computing, it’s important to understand that not all examples of this problem within the drug discovery and development process are well-suited for quantum acceleration.
Put simply, not all molecular modeling problems need higher computational accuracy, and it was critical to both the IonQ and AstraZeneca team to narrow this broad problem space into a step in the process that had the potential to genuinely solve specific, real-world computational challenges within AstraZeneca’s computational chemistry workflows. Understanding these practical use cases in detail, and designing workflows that are specifically tuned to solving them is a critical first step toward quantum value and broad quantum adoption.
It’s also important to note that this workflow is not designed or intended to replace AstraZeneca’s existing computational chemistry workflows, but rather augment, accelerate and enhance what already exists. IonQ believes that quantum computing’s benefits won’t come from replacing classical systems outright, but from accelerating critical computational bottlenecks by strategically integrating quantum in a hybrid model.
The hybrid quantum application discussed here is the result of a focused effort of IonQ application scientists working alongside AstraZeneca domain experts to address one such bottleneck, working together to provide a concrete, fully-worked example of how we expect quantum to deliver practical, near-term impact.
Background: Life Sciences Industry and Use Cases
The average cost of bringing a novel drug to market exceeds $2 billion over a 10–15 year timeline [Deloitte], with the drug development process spanning multiple stages. These stages often involve significant computational resources for tasks such as molecular simulation, optimization, and data analysis. Accelerating even portions of these workflows with quantum computing presents a promising opportunity for innovation for both AstraZeneca and IonQ.
Recent progress in quantum computing now means that practical, real-world applications are on the horizon, opening the door to faster and more innovative approaches to decades-old problems, with the drug design and development process as a key early opportunity area. In particular, quantum algorithms are beginning to show promise in addressing the computational challenges associated with predicting the properties and potential interactions of complex molecular systems with high accuracy.
We identified specific quantum chemistry challenges where quantum could deliver a meaningful advantage, with a key focus on simulating strongly correlated electrons and complex reaction mechanisms in drug candidates with high accuracy, in parts of the drug discovery and development pipeline where accuracy is currently a limiting factor.
One key opportunity area of several identified in our research was the design of chemical reactions where transition metals — metallic elements such as iridium, platinum, nickel, and copper — are used not as pharmaceuticals themselves, but as catalysts to help produce target molecules. “Catalysis is employed throughout the pharmaceutical lifecycle, including drug discovery and development, helping chemists selectively synthesize high-purity drugs. Simulations can most effectively influence timelines during the development phase, where selectivity and scalability are critical.
One example where these transition metal catalysts are especially useful are for molecules that have two chiral or “mirror image” versions, where one of these mirror images has more desirable pharmaceutical properties than the other — in extreme cases, being able to selectively make only one of the two mirror image molecules is the difference between making medicine and making poison.

Improving the ability to precisely predict the mechanisms and outcomes of these catalytic reactions allows drug designers to identify catalysts and reactions that will let them synthesize just the molecules they want in more sustainable, higher-yield, or more cost-effective ways. This has obvious benefits at full production scale, but can even provide meaningful benefits while drugs are in development. In preclinical and clinical trials, large amounts of the candidate drug need to be produced to evaluate efficacy, toxicity, and other qualities; even marginal improvements in yield can have huge impacts when literal tons of material may need to be produced even before the drug ever makes it to a production line.
These problems are particularly well suited to quantum acceleration because the computational cost of modeling these interactions at the level of accuracy necessary to predict the complicated electronic interactions that allow for “mirror image” selectivity and other very specific behaviors grows exponentially with the number of electron orbitals. This exponential scaling means that high-accuracy modeling easily surpasses the capabilities of classical computing for anything but the simplest molecules, and almost all commercially significant compounds.
By contrast, quantum computers have little trouble with this exponential behavior. With the right algorithmic techniques, quantum computers can encode an exponentially growing state space into a linearly growing number of qubits — for every N additional qubits, 2^N more states can be described and processed. This allows for the modeling of much larger systems at high fidelity than is possible with classical processors, with only modest qubit counts.
The Approach: Quantum Monte Carlo with Matchgate Shadows to Accelerate Time-to-Solution at Scale
For over a year, the IonQ team has been hyperfocused on identifying innovative algorithmic approaches that provide both near-term promise and long-term scalability. Our innovative approach, based on the Quantum-Classical Auxiliary-Field Quantum Monte Carlo (QC-AFQMC) algorithm, is one of our frontrunners in this initiative. QC-AFQMC is a well-researched technique with a variety of promising qualities, such as efficient scaling with increasing system size, inherent resilience to quantum gate noise, and the ability to further reduce quantum resource requirements by cleverly relying on a combination of quantum and classical data processing techniques, making it a viable solution for both near-term and future quantum applications.
Within the context of pharmaceutical drug synthesis, this approach can enable more accurate electronic structure modeling, providing deeper insights into catalysis and reaction mechanisms beyond the capabilities of classical methods like Density Functional Theory (DFT).
Last year, our collaborators at AWS published a research paper demonstrating a hybrid quantum-classical algorithm designed to apply the Quantum Monte Carlo algorithm to the electronic structure problem in quantum chemistry. Building upon previous work that utilized Clifford shadows, the authors employ Matchgate shadows to mitigate the exponential scaling issues associated with post-processing. Their experiments on the IonQ Aria quantum hardware demonstrate that this approach is inherently noise-robust; however, they provided a pessimistic outlook for the classical post-processing, which remained computationally intensive, posing challenges for the algorithm as it scaled into industrially-relevant problem sizes.
Our enhanced approach, as described in this blog, shows a meaningful acceleration in end-to-end time-to-solution—measured against the prior reference work by AWS—by optimizing both the quantum execution and the classical post-processing stages of the workflow with a combination of novel algorithmic techniques, hardware improvements, GPU acceleration, and tight quantum-classical integration via NVIDIA CUDA-Q and Amazon Braket.
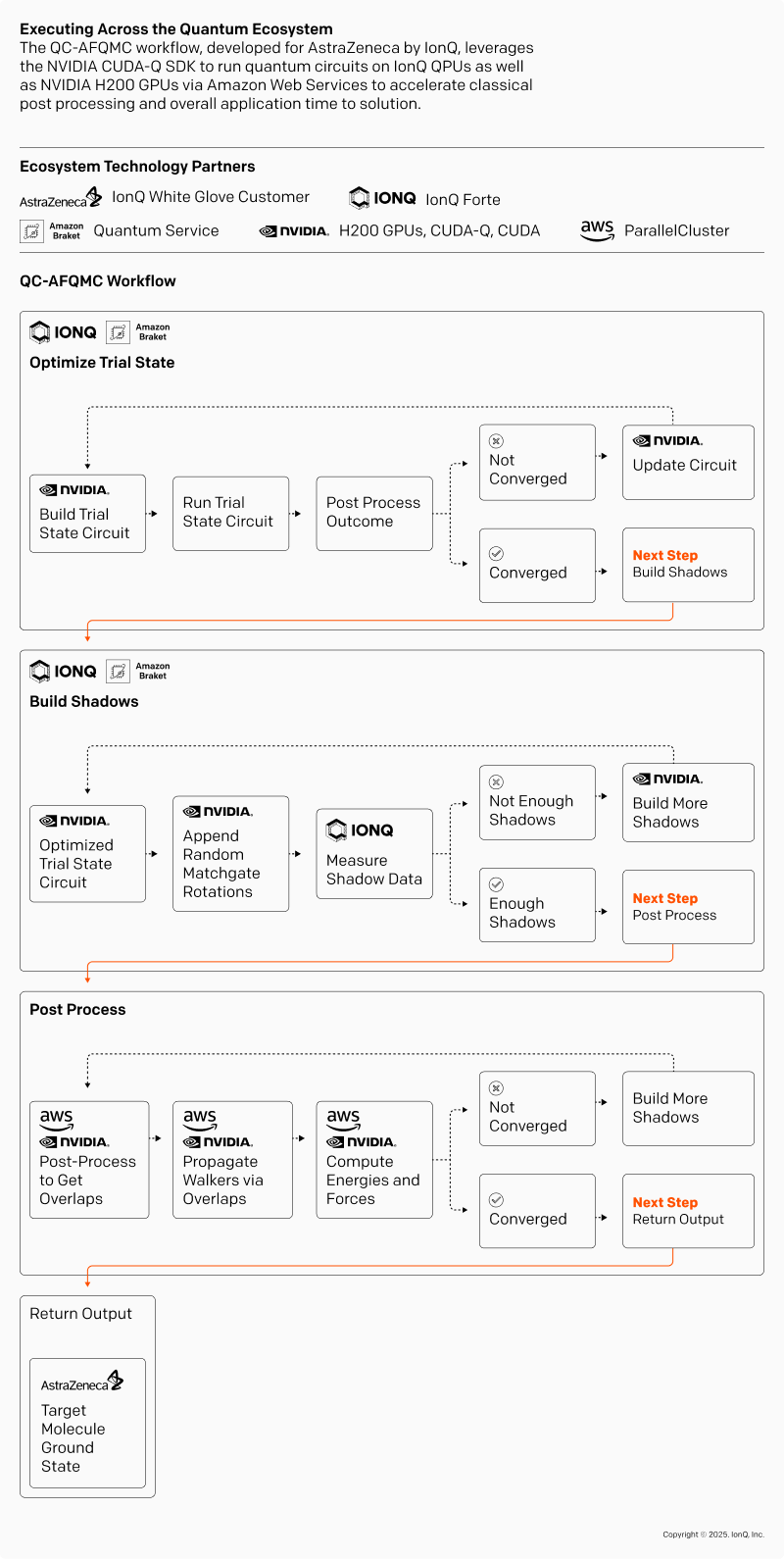
The novel IonQ-designed approach shows how quantum computing can enable QC-AFQMC calculations for modeling the Suzuki–Miyaura cross-coupling reaction with a nickel catalyst, a specific example of the transition metal enabled reactions described above. Here are the key steps of the workflow:
- Trial State Preparation: A low-cost upCCD ansatz (unitary pair coupled cluster with double excitations) was used to construct an efficient, hardware-friendly initial state (an approximation of the system's ground state), and this trial state was further optimized using a Variational Quantum Eigensolver (VQE).
- Quantum Measurement & Classical Shadow Encoding: The trial wave function was repeatedly prepared and measured on IonQ Forte (via Amazon Braket) using “matchgate shadows,” a type of shadows, which enable efficient reconstruction of observables while reducing the number of measurements needed (see visual below). We applied a custom designed error detection flags for post-selection.
- Propagation: Walkers evolved under the molecular Hamiltonian through imaginary time propagation using the trial wave function. This step helps address the phase problem, a key challenge in quantum Monte Carlo simulations.
Overlap & Energy Computation: Wave function overlaps and ground-state energies were computed using efficient matrix operations. These computations were accelerated using NVIDIA GPUs (via AWS ParallelCluster), leveraging classical resources to handle the post-processing efficiently.
.gif)
The IonQ, AstraZeneca, NVIDIA, and AWS teams further optimized the process to accelerate time-to-solution over the previous reference demonstration of this approach. The core innovations that led to the optimization of this application included:
- Matchgate Shadows: Reduced overlap computation cost, avoiding exponential scaling issues.
- GPU Acceleration: Speeded up matrix operations and reduced bottlenecks in large QC-AFQMC simulations.
- Hybrid Parallelization: Distributed tasks across multiple GPUs and CPUs, enabling concurrent execution with minimal overhead.
With these improvements, we were able to accelerate the quantum portion of the workload by 9x, running over 275,000 shadow measurements at a median duration of 1.1s per circuit, as opposed to a measured median duration of 9.9s in a comparable test with the runtime accelerations turned off. In real terms, this translated to a total runtime per molecule of about 18 hours, down from an estimated week or more. We were further able to dramatically accelerate the HPC post-processing when compared to the previous CPU-only implementation, where it was a key bottleneck. Our innovative post-processing algorithm is the main driver of the speedup. The high-performance GPU-accelerated implementation executed in the AWS ParallelCluster environment yielded an orders-of-magnitude time reduction compared to what the state-of-the-art algorithm would have taken. Because the existing algorithm is unable to work with molecules as large as the ones we used in this study, it is difficult to pinpoint the speedup exactly, however we conservatively estimate that this workload — just on benchmarked improvements — to have given us a real-world, end-to-end speedup of at least 20x, accomplishing in just a few days what would have previously taken months.
All-in, these results demonstrate the power of co-designed, integrated application workflows that can make the most of high-performance quantum hardware like IonQ’s, by aligning everything else up the stack–circuit execution runtime, the error mitigation approach, the classical postprocessing, algorithmic techniques, and more–to advance chemistry and materials science by helping to overcome classical computational limits in high-accuracy simulations and enabling analysis of more complex chemical systems.
Maximizing Customer Value With Leading Partners
Last year, we began rolling out the earliest pieces of our Hybrid Services suite within the IonQ Quantum Cloud. This suite of new features enables our customers and partners to easily provision hybrid quantum-classical infrastructure, executing quantum-accelerated workloads across IonQ systems and high-performance cloud resources. This allows applications researchers to spend more time exploring new use cases and less time fighting with infrastructure, and allows infrastructure engineers to integrate these applications into existing enterprise workflows, considerably reducing not just time-to-solution, but also overall time-to-value.
IonQ’s unique commitment to being wherever our customers are and supporting whatever tools our customers use — whether that’s any of the major public clouds, any of the major quantum SDKs — was on particular display in this work. Broad interoperability and open ecosystems have been at the core of our business and technology strategy since we launched the IonQ Quantum Cloud in 2019, and as we began identifying key use cases with the AstraZeneca team, it became clear that making it a success would require much more than what IonQ could provide alone. Our pre-existing integrations and relationships with Amazon Braket and the larger AWS ecosystem, the NVIDIA team for CUDA-Q, applications, and HPC-scale GPU acceleration insight, made it easy to bring the very best of the quantum ecosystem to bear on this complex application. Leveraging the IonQ Hybrid Services suite as our core orchestration engine, our applications team were able to use these best-in-class tools and call upon leading experts to integrate and optimize every piece of this workload, ultimately achieving major improvements in application performance and time-to-solution.
The hybrid quantum-classical pipeline assembled around IonQ Forte includes:
- Quantum Processing Unit (QPU): IonQ Forte
- Quantum Development Platform: NVIDIA CUDA-Q
- Workflow Orchestration: Amazon Braket (AWS)
- Classical Processing: NVIDIA GPUs (H200s via P5en instances on AWS ParallelCluster)
The end-to-end QC-AFQMC workflow was executed leveraging core capabilities and strengths across this tech stack, with the goal of modeling a nickel-catalyzed chemical reaction of interest to AstraZeneca for its applications in drug design and synthesis.
Summary and Path Forward
This demonstration represents both powerful ecosystem collaboration and a meaningful step towards practical quantum computing applications in chemistry and materials science. By successfully integrating quantum and classical computing at scale with a novel QC-AFQMC approach, our collaboration with NVIDIA, AstraZeneca, and AWS showcases the tangible benefits of quantum acceleration for complex simulations. The ability to model catalytic reactions with speed and accuracy isn’t just an exciting scientific achievement—it’s a preview of how hybrid computing and quantum acceleration will provide revolutionary capabilities to the industry.
We are excited to uncover other commercial opportunities for near-term quantum computing as we continue to partner with our partners and customers around the world. With a flexible software stack and the demonstrated ability to execute customer focused workflows across the ecosystem, we’re only beginning to unlock quantum’s full potential.
Read the paper on arXiv.

